EinleitungMedizinprodukte spielen eine wesentliche Rolle in der Gesundheitsversorgung und können einen erheblichen Einfluss auf die Gesundheit und Sicherheit der Patienten haben. Um die Sicherheit und Wirksamkeit von Medizinprodukten zu gewährleisten, verlangen die Aufsichtsbehörden weltweit von den Herstellern die Durchführung einer Risikobewertung und einer biologischen Sicherheitsbewertung. Dieser Bewertungsprozess umfasst die Erstellung eines biologischen Bewertungsplans(BEP) und die Vorlage eines biologischen Bewertungsberichts(BER), der auch als biologische Risikobewertung(BRA) bezeichnet wird.Was ist ein biologischer Bewertungsplan (BEP) und warum ist er notwendig?Der BEP ist ein wichtiges Dokument, das die Strategie und Methodik der biologischen Sicherheitstests für ein Medizinprodukt beschreibt. Er wird auf der Grundlage der Norm 10993-1 der Internationalen Organisation für Normung (ISO) erstellt, die Leitlinien für die biologische Bewertung von Medizinprodukten enthält. Der BEP enthält in der Regel eine Beschreibung des Produkts, seiner Zweckbestimmung und der Zielpatientengruppe. Die Risikobewertung und der Plan für die Durchführung der biologischen Sicherheitstests werden ebenfalls im BEP beschrieben. Der BEP wird den zuständigen Behörden als Teil des Zulassungsantrags für das Produkt vorgelegt.Welche Parameter sollten für den BEP in Betracht gezogenwerden? Bei der Bewertung von Materialien oder Medizinprodukten, die für den menschlichen Gebrauch bestimmt sind, muss ein biologischer Bewertungsplan Teil eines strukturierten Risikomanagementprozesses gemäß ISO 14971:2007, Anhang I, sein.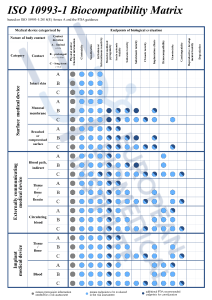 Abbildung 1. Endpunkte, die bei einer biologischen Risikobewertung zu berücksichtigen sind.1,3Dieser Prozess umfasst die Identifizierung biologischer Gefahren, die Bewertung der damit verbundenen Risiken und die Bestimmung ihrer Akzeptanz. Anhang B enthält eine Anleitung für diesen Prozess. Die Bewertung muss von sachkundigen und erfahrenen Fachleuten geplant, durchgeführt und dokumentiert werden: „Die Bewertung muss eine dokumentierte, fundierte Abwägung der Vorteile/Nachteile und der Relevanz folgender Punkte umfassen
Abbildung 1. Endpunkte, die bei einer biologischen Risikobewertung zu berücksichtigen sind.1,3Dieser Prozess umfasst die Identifizierung biologischer Gefahren, die Bewertung der damit verbundenen Risiken und die Bestimmung ihrer Akzeptanz. Anhang B enthält eine Anleitung für diesen Prozess. Die Bewertung muss von sachkundigen und erfahrenen Fachleuten geplant, durchgeführt und dokumentiert werden: „Die Bewertung muss eine dokumentierte, fundierte Abwägung der Vorteile/Nachteile und der Relevanz folgender Punkte umfassen
- a) Konfiguration des Medizinprodukts (z. B. Größe, Geometrie, Oberflächenbeschaffenheit) und Auflistung der Konstruktionsmaterialien eines Medizinprodukts (qualitativ) und, falls erforderlich, des Anteils und der Menge (Masse) jedes Materials in dem Medizinprodukt (quantitativ);
- b) die physikalischen und chemischen Eigenschaften der verschiedenen Konstruktionswerkstoffe und ihre Zusammensetzung;
- c) etwaige Daten über die klinische Verwendung oder die Exposition des Menschen;
- d) alle vorhandenen toxikologischen und sonstigen biologischen Sicherheitsdaten über Produkt und Bestandteile, Abbauprodukte und Metaboliten;
- e) Prüfverfahren. „1
Im Risikomanagementplan sollte angegeben werden, welche technischen Kompetenzen für die biologische Bewertung erforderlich sind und wer dafür verantwortlich ist. Bei der Bewertung sollten die Vor- und Nachteile des Materials oder Produkts und ihre Relevanz berücksichtigt werden, und diese Erwägung sollte dokumentiert werden. „4.3 Die folgenden Punkte sind aufgrund ihrer Relevanz für die biologische Gesamtbewertung des Medizinprodukts zu berücksichtigen
- a) das bzw. die Konstruktionsmaterial(ien) (d. h. alle direkt und indirekt gewebeberührenden Materialien);
- b) vorgesehene Zusatzstoffe, Prozessverunreinigungen und Rückstände (z. B. ist die Prüfung auf Ethylenoxid-Sterilisationsrückstände gemäß ISO 10993-7 durchzuführen);
- c) Verpackungsmaterialien, die direkt oder indirekt mit dem Medizinprodukt in Berührung kommen, können Chemikalien auf das Medizinprodukt und dann indirekt auf den Patienten oder Kliniker übertragen;
- d) auslaugbare Stoffe (siehe ISO 10993-17 und ISO 10993-18);
- e) Abbauprodukte (siehe ISO 10993-9 für allgemeine Grundsätze und 10993-13, 10993-14 und 10993-15 für Abbauprodukte aus Polymeren, Keramiken bzw. Metallen);
- f) andere Bestandteile und ihre Wechselwirkungen im Endprodukt;
- g) die Leistung und die Eigenschaften des Endprodukts;
- h) physikalische Eigenschaften des Endprodukts, einschließlich, aber nicht beschränkt auf, Porosität, Partikelgröße, Form und Oberflächenmorphologie „1
Bevor biologische Tests an einem Medizinprodukt durchgeführt werden, müssen seine chemischen Bestandteile beschrieben und seine Materialien charakterisiert werden, einschließlich der chemischen Charakterisierung gemäß ISO 10993-18. Die chemische Charakterisierung kann dabei helfen festzustellen, ob weitere Tests unter Verwendung eines geeigneten toxikologischen Schwellenwerts erforderlich sind. Anhang B, ISO 10993-17 und ISO 10993-18 enthalten Anleitungen zu diesem Prozess. Warum ist der Biologische Bewertungsbericht (BER) so wichtig?Der BER ist ein Dokument , das die Ergebnisse der biologischen Sicherheitsprüfung eines Medizinprodukts zusammenfasst. Die Tests werden auf der Grundlage der BEP- und ISO 10993-1-Richtlinien durchgeführt. Die GVO enthält eine Zusammenfassung der Prüfmethoden und -ergebnisse, eine Bewertung der bei der Prüfung festgestellten potenziellen Risiken und eine Schlussfolgerung zur Sicherheit des Produkts auf der Grundlage der Prüfergebnisse. Der BER wird auch den zuständigen Behörden als Teil des Zulassungsantrags für das Produkt vorgelegt. Verfassen eines ausgezeichneten biologischen Bewertungsberichts (BER) auf der Grundlage der Erfahrungen der Industrie:
Warum ist der Biologische Bewertungsbericht (BER) so wichtig?Der BER ist ein Dokument , das die Ergebnisse der biologischen Sicherheitsprüfung eines Medizinprodukts zusammenfasst. Die Tests werden auf der Grundlage der BEP- und ISO 10993-1-Richtlinien durchgeführt. Die GVO enthält eine Zusammenfassung der Prüfmethoden und -ergebnisse, eine Bewertung der bei der Prüfung festgestellten potenziellen Risiken und eine Schlussfolgerung zur Sicherheit des Produkts auf der Grundlage der Prüfergebnisse. Der BER wird auch den zuständigen Behörden als Teil des Zulassungsantrags für das Produkt vorgelegt. Verfassen eines ausgezeichneten biologischen Bewertungsberichts (BER) auf der Grundlage der Erfahrungen der Industrie:
- Beginnen Sie mit dem BER schon früh im Entwicklungsprozess und nutzen Sie ihn als lebendes Dokument, um potenzielle Lücken in der biologischen Sicherheit des Produkts zu ermitteln.
- Identifizieren Sie die mit dem Produkt verbundenenRisiken, bevor Sie mit dem Bewertungsprozess beginnen, und dokumentieren Sie alle relevanten Details.
- Arbeiten Sie mit Experten zusammen, die über die technischen Fähigkeiten und die Ausbildung verfügen, um die erforderlichen Richtlinien zu interpretieren und korrekt anzuwenden.
- Überprüfen Sie die GVO jährlich, um Möglichkeiten zur Erhöhung der Sicherheit oder Änderungen, die sich auf die Sicherheit des Produkts auswirken könnten, zu ermitteln.
Zu den häufigen Fehlern, die es zu vermeiden gilt, gehören eine unzureichende Datenerfassung, die Nichtberücksichtigung aller Materialien mit Patientenkontaktund die Auswahl von Materialien nach dem Preis. Die Risikobewertung ist ein wesentliches Element eines gründlichen biologischen Bewertungsberichts. Das mit dem Medizinprodukt verbundene Risiko steigt mit dem Ausmaß und der Invasivität des Patientenkontakts, und es ist wichtig, eine Hierarchie der Risiken und die entsprechenden Bewertungskriterien zu dokumentieren.Was regelt die ISO 10993-1?Die ISO 10993-1 ist die wichtigste Norm zur Bewertung der biologischen Sicherheit von Medizinprodukten. Sie bietet einen Leitfaden für die Risikobewertung und die Beurteilung der biologischen Sicherheit von Medizinprodukten. Die ISO-Norm 10993 umreißt die verschiedenen Tests, die zur Bewertung der biologischen Sicherheit eines Medizinprodukts durchgeführt werden sollten. Dazu gehören Tests für physikalische und/oder chemische Informationen, Zytotoxizität, Sensibilisierung, Reizung oder intrakutane Reaktivität, materialvermittelte Pyrogenität, akute systemische Toxizität, subakute Toxizität, subchronische Toxizität, chronische Toxizität, Implantationseffekte, Genotoxizität, Karzinogenität, Reproduktions-/Entwicklungstoxizität und Degradation. Die Einhaltung der ISO 10993 wird von Regulierungsbehörden auf der ganzen Welt gefordert, darunter auch von der FDA. ISO 10993-1 enthält die folgenden Leitlinien für die Regulierung von Studien über Medizinprodukte:
- Allgemeine Grundsätze für die biologische Bewertung von Medizinprodukten
- Kategorisierung von Medizinprodukten
- Prozess der biologischen Bewertung
- Interpretation der biologischen Bewertungsdaten und allgemeine biologische Risikobewertung
Empfehlungen der FDADie FDA ist in den Vereinigten Staaten für die Regulierung von Medizinprodukten zuständig. Im Rahmen des Zulassungsverfahrens müssen die Hersteller einen Zulassungsantrag einreichen, der einen BEP und eine BER enthält. Die FDA prüft diese Dokumente, um sicherzustellen, dass das Produkt für die vorgesehene Verwendung sicher und wirksam ist. Die Nichteinhaltung der FDA-Vorschriften kann zu erheblichen Verlusten führen, z. B. zu Rückrufaktionen: „Ein solcher Prozess sollte im Allgemeinen mit der Bewertung des Produkts beginnen, einschließlich der Materialkomponenten, der Herstellungsprozesse und derklinischen Verwendung des Produkts (…) Unter Berücksichtigung dieser Informationen sollten die potenziellen Risiken aus Sicht der Biokompatibilität ermittelt werden.
Unter Berücksichtigung der potenziellen biologischen Auswirkungen sollte ein Plan entwickelt werden (…) entweder durch Biokompatibilitätstests oder andere Bewertungen, die die Risiken angemessen berücksichtigen. „2MDR (Medical Device Regulation) RichtlinienDie MDR ist ein Regelwerk, das die Zulassung und Verwendung von Medizinprodukten in der Europäischen Union regelt. Die MDR schreibt vor, dass die Hersteller von Medizinprodukten eine biologische Sicherheitsbewertung nach dem neuesten Stand der Technik durchführen müssen, was bedeutet, dass die Bewertung auf dem neuesten Stand der wissenschaftlichen Erkenntnisse und der Technik erfolgen muss. Die Bewertung muss auch die bestimmungsgemäße Verwendung des Produkts und alle potenziellen Risiken, die mit seiner Verwendung verbunden sind, berücksichtigen. Darüber hinaus müssen die Hersteller einen BEP und eine BER als Teil des Zulassungsantrags vorlegen. Die MDR enthält auch strengere Anforderungen an die biologische Sicherheitsbewertung von Medizinprodukten als die vorherige Medizinprodukterichtlinie (MDD).Bedeutung von ExpertenDie biologische Sicherheitsbewertung ist ein komplexer Prozess, der das Fachwissen ausgebildeter Fachleute erfordert. Daher ist es unerlässlich, Experten in die Entwicklung der BEP und BER einzubeziehen, um sicherzustellen, dass die Tests korrekt geplant und die Ergebnisse richtig interpretiert werden. Experten können auch bei der Auswahl geeigneter Prüfmethoden und der Identifizierung potenzieller Risiken im Zusammenhang mit dem Produkt behilflich sein. „(…) Die biologische Bewertung muss von sachkundigen und erfahrenen Fachleuten geplant, durchgeführt und dokumentiert werden.(…) „1 SchlussfolgerungDie biologische Sicherheitsbewertung von Medizinprodukten ist ein entscheidender Schritt zur Gewährleistung der Patientensicherheit und der Einhaltung von Vorschriften. Die BEP und BER sind wichtige Dokumente, die die Teststrategie umreißen und die Ergebnisse der biologischen Sicherheitsbewertung zusammenfassen. Für die Vermarktung eines Medizinprodukts ist die Einhaltung der ISO 10993, der FDA und der MDR-Vorschriften erforderlich. Es ist wichtig, Experten in den Prozess der biologischen Sicherheitsbewertung einzubeziehen, um sicherzustellen, dass die Tests korrekt durchgeführt und die Ergebnisse richtig interpretiert werden, wie es in der ISO 10993-1:2018 heißt. Zusammenfassend lässt sich sagen, dass der biologische Bewertungsplan und der Bericht entscheidende Dokumente im behördlichen Zulassungsverfahren für Medizinprodukte sind. Die Einhaltung der ISO 10993, der FDA und der MDR-Vorschriften ist Voraussetzung für die Vermarktung eines Medizinprodukts. Der BEP und der BER dienen als Fahrplan und Zusammenfassung des biologischen Sicherheitsbewertungsprozesses für ein Medizinprodukt, und die Einbeziehung von Experten in den Prozess ist unerlässlich, um seine Genauigkeit und Zuverlässigkeit zu gewährleisten. Durch die Einhaltung der von den Aufsichtsbehörden festgelegten Richtlinien und Normen und die Einbeziehung von Experten in den Prozess können die Hersteller sicherstellen, dass ihre Medizinprodukte sicher und wirksam für den Einsatz beim Patienten sind. Referenzen:
- ISO 10993-1:2018 Biologische Beurteilung von Medizinprodukten – Teil 1: Beurteilung und Prüfung im Rahmen eines Risikomanagementprozesses
- Grundlagen der Biokompatibilität: Erforderliche Informationen für die Bewertung durch die FDA
- Verwendung der internationalen Norm ISO 10993-1, „Biologische Beurteilung von Medizinprodukten – Teil 1: Beurteilung und Prüfung im Rahmen eines Risikomanagementprozesses“.
