The Importance of BEP and BER
The Importance of Biological Evaluation Plan and Report in Medical Devices
Authors: Bartosz Gromadka1, Łukasz Szymański1, Damian Matak1
1 European Biomedical Institute, Józefów, Poland
* info@ebi.bio; https://ebi.bio/
Keywords: biocompatibility, ISO 10993, medical device, BEP, BER, BRA
Introduction
Medical devices play an essential role in healthcare and can have a significant impact on patient health and safety. To ensure the safety and effectiveness of medical devices, regulatory authorities worldwide require manufacturers to conduct a risk assessment and biological safety evaluation. This evaluation process involves the creation of a Biological Evaluation Plan (BEP) and the submission of a Biological Evaluation Report (BER), also known as a Biological Risk Assessment (BRA).
What is Biological Evaluation Plan (BEP), and why is it necessary?
The BEP is a critical document that outlines the biological safety testing strategy and methodology for a medical device. It is developed based on the International Organization for Standardization (ISO) 10993-1 standard, which provides guidance on the biological evaluation of medical devices. The BEP typically includes a description of the device, its intended use, and the target patient population. The risk assessment and plan for conducting biological safety testing are also outlined in the BEP. The BEP is submitted to competent authorities as part of the device’s regulatory submission.
What parameters are worth considering for BEP?
When evaluating any material or medical device intended for human use, a biological evaluation plan must be part of a structured risk management process according to ISO 14971:2007, Annex I.
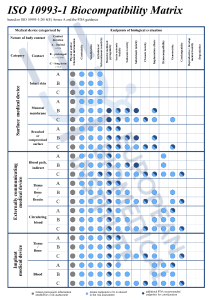
Figure 1. Endpoints to be addressed in a biological risk assessment.1,3
This process involves identifying biological hazards, assessing the associated risks, and determining their acceptability. Annex B provides guidance for this process. The evaluation must be planned, conducted, and documented by knowledgeable and experienced professionals.
“The evaluation shall include documented, informed consideration of advantages/disadvantages and relevance of:
a) medical device configuration (e.g. size, geometry, surface properties) and a listing of a medical device’s materials of construction (qualitative) and where necessary, the proportion and amount (mass) of each material in the medical device (quantitative);
b) the physical and chemical characteristics of the various materials of construction and their composition;
c) any history of clinical use or human exposure data;
d) any existing toxicology and other biological safety data on product and component materials, breakdown products and metabolites;
e) test procedures.”1
The risk management plan should specify which technical competencies are required for the biological evaluation and identify who is responsible for it. The evaluation should consider the advantages and disadvantages of the material or device and its relevance, and this consideration should be documented.
“4.3 The following shall be taken into account for their relevance to the overall biological evaluation of the medical device:
a) the material(s) of construction (i.e. all direct and indirect tissue contacting materials);
b) intended additives, process contaminants and residues (for example, testing for ethylene oxide sterilization residuals shall be conducted in accordance with ISO 10993-7);
c) packaging materials that directly or indirectly contact the medical device can transfer chemicals to the medical device and then indirectly to the patient or clinician;
d) leachable substances (see ISO 10993-17 and ISO 10993-18);
e) degradation products (see ISO 10993-9, for general principles and 10993-13, 10993-14 and 10993-15 for degradation products from polymers, ceramics and metals, respectively);
f) other components and their interactions in the final product;
g) the performance and characteristics of the final product;
h) physical characteristics of the final product, including but not limited to, porosity, particle size, shape and surface morphology”1
Before conducting any biological testing on a medical device, its chemical constituents must be described and its materials must be characterized, including chemical characterization in accordance with ISO 10993-18. Chemical characterization can help determine if further testing is necessary, using an appropriate toxicological threshold. Annex B, ISO 10993-17, and ISO 10993-18 provide guidance on this process
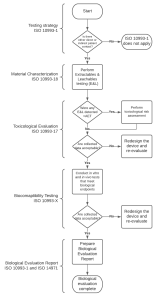
Figure 2. Flowchart diagram of biocompatibility assessment.
Why is Biological Evaluation Report (BER) so important?
The BER is a document that summarizes the results of the biological safety testing performed on a medical device. The testing is conducted based on the BEP and ISO 10993-1 guidelines. The BER includes a summary of the test methods and results, an evaluation of any potential risks identified during testing, and a conclusion on the device’s safety based on the results of the testing. The BER is also submitted to competent authorities as part of the device’s regulatory submission.
Writing an excellent Biological Evaluation Report (BER) based on industry experience:
-
Begin the BER early in the development process and use it as a living document to identify potential gaps in the biological safety of the device.
-
Identify the risks associated with the device before starting the evaluation process and document all relevant details.
-
Work with experts who have the technical skills and training to interpret the necessary guidelines and apply them correctly.
-
Revisit the BER annually to identify opportunities for increased safety or changes that may impact the product’s safety.
Some common mistakes to avoid include poor data collection, failing to consider all materials with patient contact, and choosing materials by price.
Risk assessment is a crucial element of a thorough biological evaluation report. The level of risk associated with the medical device increases with the extent and invasiveness of patient contact, and it’s important to document a hierarchy of risks and corresponding evaluation criteria.
What is regulated by ISO 10993-1?
ISO 10993-1 is the primary standard used to evaluate the biological safety of medical devices. It provides guidance on the risk assessment and biological safety evaluation of medical devices. The ISO 10993 standard outlines the various tests that should be conducted to assess the biological safety of a medical device, including tests for physical and/or chemical information, cytotoxicity, sensitization, irritation or intracutaneous reactivity, material-mediated pyrogenicity, acute systemic toxicity, subacute toxicity, subchronic toxicity, chronic toxicity, implantation effects, genotoxicity, carcinogenicity, reproductive/developmental toxicity, and degradation. Compliance with ISO 10993 is required by regulatory authorities worldwide, including the FDA.
ISO 10993-1 gives the following guidelines for regulating studies on medical devices:
-
General principles applying to biological evaluation of medical devices
-
Categorization of medical devices
-
Biological evaluation process
-
Interpretation of biological evaluation data and overall biological risk assessment
Recommendations of the FDA
The FDA is responsible for regulating medical devices in the United States. As part of the regulatory process, manufacturers should submit a regulatory submission that includes a BEP and BER. The FDA reviews these documents to ensure that the device is safe and effective for its intended use. Failure to comply with FDA regulations can result in significant losses, such as recalls.
“Such a process should generally begin with assessment of the device, including the material components, the manufacturing processes, the clinical use of the device (…) Considering this information, the potential
risks from a biocompatibility perspective should be identified. Considering the potential biological impact, a plan should be developed (…) either by biocompatibility testing or other evaluations that appropriately address the risks.”2
MDR (Medical Device Regulation) guidelines
The MDR is a set of regulations that governs the approval and use of medical devices in the European Union. The MDR requires that medical device manufacturers conduct a biological safety evaluation according to the state-of-the-art guidelines, which means that the evaluation must use the latest available scientific knowledge and technology. The evaluation must also take into account the intended use of the device and any potential risks associated with its use. In addition, it requires manufacturers to submit a BEP and BER as part of the regulatory submission. The MDR also includes stricter requirements for the biological safety evaluation of medical devices than the previous Medical Device Directive (MDD).
Importance of Experts
Biological safety evaluation is a complex process that requires the expertise of trained professionals. Therefore, it is essential to involve experts in the development of the BEP and BER to ensure that the testing is planned correctly and that the results are interpreted accurately. Experts can also provide guidance on the selection of appropriate test methods and the identification of potential risks associated with the device.
“(…) The biological evaluation shall be planned, carried out, and documented by knowledgeable and experienced professionals. (…)”1
Conclusion
The biological safety evaluation of medical devices is a critical step in ensuring patient safety and regulatory compliance. The BEP and BER are essential documents that outline the testing strategy and summarize the results of the biological safety evaluation. Compliance with ISO 10993, FDA, and MDR regulations is required to market a medical device. It is important to involve experts in the biological safety evaluation process to ensure that the testing is conducted accurately and that the results are interpreted correctly, as it is written in ISO 10993-1:2018.
In conclusion, the biological evaluation plan and report are critical documents in the regulatory approval process of medical devices. Compliance with ISO 10993, FDA, and MDR regulations is required to market a medical device. The BEP and BER serve as a roadmap and summary of the biological safety evaluation process for a medical device, and involving experts in the process is essential to ensure its accuracy and reliability. By following the guidelines and standards set forth by regulatory authorities and involving experts in the process, manufacturers can ensure that their medical devices are safe and effective for patient use.
References:
-
ISO 10993-1:2018 Biological evaluation of medical devices — Part 1: Evaluation and testing within a risk management process
-
Basics of Biocompatibility: Information Needed for Assessment by the FDA
-
Use of International Standard ISO 10993-1, “Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process”